주요질환
- 현재 페이지경로
-
- HOME
- 소아신경질환 바로알기
- 주요질환
프라더윌리 증후군 (Prader-Willi syndrome)
- 개요
-
프라더-윌리 증후군(Prader-Willi syndrome)은 1956년 Prader, Labhart, Willi에 의해 처음 보고된 유전질환으로 15번 염색체에 이상으로 발생합니다.
출생아 10,000∼15,000명 중 1명의 빈도로 발생하며, 남녀의 발생 비율은 비슷합니다. 특징적으로 작은 키와 비만, 과도한 식욕, 근육긴장 저하, 성선기능 저하증, 지적장애 등이 있습니다. 영아기에는 근육긴장 저하, 수유곤란, 성장장애가 나타나며, 아동기에 접어들면서 작은 키, 성기 발육부전 등 성적 기능의 장애, 과도한 식욕이 특징적으로 나타납니다. ‘과도한 식욕’은 시상하부의 기능이 저하되는 것과 몸에서 대사가 원활하지 않은 낮은 대사율에 의해 식욕이 증가되고 포만감이 결여되어 나타나는 증상으로 비만을 초래하게 됩니다. 비만을 조절 하지 못하여 대체로 당뇨병, 고혈압, 뇌혈관 질환 등 생명을 위협하는 심각한 합병증이 생길 수 있습니다.
Prader-Willi 증후군은 아버지로부터 유래한 15번 염색체의 이상이 주원인으로서 크게 3가지로 분류할 수 있습니다.
첫째, 아버지로부터 유래한 15번 염색체의 장완 근위부(15q11q13)의 미세결실이 원인인 경우가 75% 정도를 차지하며 대부분 자연발생적으로 생겨납니다.
둘째, 부모로부터 각각 한 개씩 유전되어야 할 15번 염색체 두 개가 모두 어머니로부터만 유래되는 uniparental disomy (UPD)로 인해 Prader-Willi 증후군이 되는 경우로서, 25%를 차지합니다.
셋째, 나머지 드문 원인으로는 15번 염색체의 장완 근위부의 각인(imprinting) 부위의 유전자에서 돌연변이가 발생한 경우입니다.
- 증상 및 징후
-
Prader-Willi 증후군은 일반적으로 유사한 특징을 가지고 있지만, 연령이 증가함에 따라 임상적인 증상과 경과가 변화합니다.
1. 신생아기
신생아 시기에는 근긴장저하로 인해 출생 전부터 태동이 저하되고, 출생 후 활동성이 축 늘어져 있으며 출생 초기에는 젖을 잘 빨지 못하고, 먹는 일에 관심을 보이지 않아 몇 주 또는 몇 달 동안 수유를 할 수 있도록 도와주어야 합니다. 그리고 울음이 약하거나 거의 울지 않습니다. 근긴장저하로 인한 수유 곤란, 성장 부진은 생후 12 개월 전에 대부분 회복됩니다. 얼굴의 특징은 좁은 이마, 아몬드 모양의 눈, 아래로 쳐진 적운세모형의 입 모양, 작은 턱 등이며, 손과 발도 작은 편입니다. 남아인 경우 음경이나 고환이 작고 잠복고환이 있으며, 여아인 경우 소음순과 음핵이 작은 것이 특징입니다.
2. 영아기
근긴장 저하로 젖이나 우유 빨기를 힘들어하며, 1살 이전까지 잘 먹으려 하지 않으므로 체중이 잘 늘지 않습니다. 다른 정상 아동과 비교하면 발달 지연을 보입니다. 근육 긴장의 저하는 구강 운동 기술과 언어발달에 영향을 주어 발음 장애가 생기고, 언어표현이 지연됩니다.
3. 아동기
2∼3세에는 식욕이 증가되면서 몸무게가 과도하게 증가되는 특징을 보입니다. 음식에 대해 끊임없이 갈망하게 되면서 음식을 훔치고 숨기는 행동을 보입니다. 강박적인 경향이 있어서 자신의 피부를 심하게 뜯어서 상처를 만들기도 하며 사소한 좌절에도 분노 발작과 문제 행동을 나타냅니다. Prader-Willi 증후군의 70~100% 에서 2세경부터 욕구불만과 관련된 행동장애를 보이기 시작 합니다. 또한 수면장애, 수면 시 무호흡이 나타날 수 있습니다. 환자의 대부분에서 지능 저하가 보이며, 지능지수는 IQ 45∼105의 범위를 나타내며 평균 지능지수는 65 정도입니다. 여러 면에서 학습장애를 보이며 학습의욕도 떨어지는 편입니다.
4. 성인기
비만과 관련되어 수면장애, 수면 시 무호흡이 나타날 수 있습니다. 아동기에 나타난 문제들이 성인기에도 계속되어 나타날 수 있습니다. 성선자극 호르몬의 분비가 정상적으로 이루어지지 않아 남녀 모두에게 사춘기가 늦게 오거나 오지 않을 수도 있고, 불임인 경우도 있습니다. 90% 의 환자에서 저신장과 골연령의 지연이 나타납니다. 출생 시 신장과 성장은 거의 정상이나 점차 성장 속도가 감소되고, 사춘기 급성장이 없어 저신장증이 나타납니다. 성인 Prader-Willi 증후군 남성의 평균 키는 155 cm, 여성의 평균 키는 148 cm 입니다.
- 진단
-
특징적인 외모와 나타나는 임상증상을 통하여 Prader-Willi 증후군을 의심해 볼 수 있으며, 분자유전학 검사나 세포유전학 검사를 통해 진단 할 수 있습니다.
염색체검사로서 15번 염색체의 장완 근위부(15q11q13)의 미세결실을 확인합니다. 15q11q13에 존재하는 SNRPN과 같은 유전자 소식자를 이용한 형광동소보합 (FISH) 검사로서 미세결실을 관찰합니다. 이 외에 uniparental disomy(UPD), 각인센터 유전자에 대한 돌연변이 검사를 할 수도 있습니다. DNA methylation 검사는 부계 유전자 결실, 2 개의 모계 유전자 존재, 유전자 각인센터의 돌연변이를 찾아낼 수 있어서 99% 정도의 Parder-Willi 증후군 환자들을 진단할 수 있습니다.
- 치료 및 예후
-
현재 특별한 치료방법은 존재하지 않지만 조기에 발견할수록 예후가 좋기 때문에 조기 진단이 중요합니다.
또한 유전상담을 통하여 적절한 정보를 제공함으로써 증상을 경감시킬 뿐만 아니라 여러 합병증을 예방할 수 있습니다. 영유아시기에는 적절한 영양 공급을 위해 특수 젖병이나, 경관 영양이 필요할 수 있습니다. 소아기에는 체중 조절을 위해 음식 섭취를 적절히 제한하고, 활동을 격려해야 합니다. 성장을 촉진 시키고, 지방 비율을 줄이기 위한 성장 호르몬 치료, 발달 재활 서비스, 조기 성숙증, 당뇨, 갑상선저하증을 위한 치료, 행동장애와 안과적 증상, 수면 장애, 경련 증상에 대한 표준 치료, 저 골밀도를 치료하기 위한 비스포스보네이트, 성장 호르몬 치료가 도움이 될 수 있습니다. 성인기에는 체중 조절을 통해 비만을 예방하고, 성장 호르몬 치료를 통해 근육량을 유지하는 것이 도움이 됩니다.
레트 증후군 (Rett syndrome)
- 개요
-
레트 증후군(Rett Syndrome)은
생후 6개월에서 18개월까지 비교적 정상 발달을 한 후 두위 발달의 감소(후천성 소두증)와 함께 습득했던 인지 및 운동 능력의 상실(퇴행), 언어기능의 상실, 그리고 손을 씻는 듯한 동작을 반복하는 특징적인 손의 상동증을 보이는 X염색체 우성으로 유전되는 질환입니다.
가족력은 드물고 여아에게 주로 나타나며, 드물게 남아에게도 나타나는 신경계 발달 질환입니다. 민족과 인종 차이 없이 여아 10,000∼15,000명당 1명으로 발생빈도가 비교적 높은 증후군으로 자폐, 뇌성마비, 규정되지 않은 발달지체로 진단이 늦어지는 경우가 많습니다.
원인은 아직 확실하게 알려지지 않았지만 X염색체의 장완에 위치(Xq28)하고 있는 MeCP2(Methyl CpG binding Protein 2) 유전자의 돌연변이에 의해 이 질환이 생길 수 있다는 것이 밝혀졌습니다. 이 유전자에 의해 만들어지는 단백질은 X 염색체 불활성화에 관여하여 결과적으로 감각, 감정, 운동신경과 자율신경의 기능을 담당하는 뇌의 특정영역의 정상적 발달에 필요한 어떤 특정 요소의 부족이나 부재의 결과를 초래합니다. 그 요소가 뇌의 발달에 필요하게 되기 전인 영아기(early infancy)에는 발육이 정상인 것으로 보이지만 이 요소가 없으면 뇌의 특정 부분이 발달상 미성숙하게 남게 되는 것이고, 이런 이유로 태어나서 첫 몇 달 동안은 아이가 정상적으로 발육하는 것처럼 보이는 것입니다. 그러나 가족력과 연관되어 유전되기보다는 대부분 가족력이 없이 산발적 돌연변이로 발생합니다.
- 증상 및 징후
-
레트 증후군의 환아는 보통 건강하게 태어나서 1세 후에 증상이 나타나기 시작합니다.
증상으로는 자신이 하고자 하는 대로 손을 자유롭게 움직이지 못하며 전형적으로 반복적이고 무의미한 손놀림이 나타나는데, 주로 양손을 씻는 듯한 동작, 비틀기, 손뼉 치기 등의 동작을 보입니다. 또한 다리에 경직감과 경련성 근육 움직임이 나타나고, 넓은 폭으로 걷는 이상한 걸음걸이 형태의 조화운동불능이 나타납니다. 머리 둘레의 성장 속도가 둔화되고, 자폐증과 같은 행동이 나타나 사람들과의 관계가 멀어지며, 의사소통이 어려워집니다. 불규칙적인 호흡을 보이기도 하며, 음식을 먹고 삼키는데 어려움을 겪습니다. 또한 성장 지연이 일어나며, 뇌에서 전기적인 활동이 통제되지 않아 발작이 나타나기도 합니다. 그 외에도 보행 장애, 다리의 경축, 운동실조의 신경학적 증상이 나타날 수 있습니다. 발작은 일반적으로 2~4세에서 시작되며, 전신강직간대발작, 단순 혹은 복합부분발작 등이 흔합니다. 레트 증후군의 모든 아동들이 이 증상 모두를 보이는 것은 아니며, 개개의 증상은 그 심각성이 다를 수 있습니다.
임상병기에 따라 1기에서 4기로 병이 진행하며
1기는 발달의 정지, 퇴행이 시작되는 시기이고, 제2기는 급속하게 발달 퇴행이 진행하는 시기로 이 시기에 진단되는 경우가 많습니다. 3기에 접어들면 퇴행의 속도가 늦춰지지만, 간질(뇌전증) 등의 합병증이 동반될 때가 많습니다. 4기가 되면 말기로 환자의 많은 수가 보행을 할 수 없게 됩니다.
- 진단
-
임상적으로 병이 의심된다면 임상 진단 기준을 참조하여 임상 진단을 내리고 유전자 검사로 확진할 수 있습니다.
레트증후군 환자의 70~80%가 위에서 언급한 MeCP2 유전자의 이상을 보여 확진할 수 있지만, 일부에서는 유전자 검사에서 음성을 보이기도 합니다. 이럴 때에는 임상진단 기준에 따른 진단을 할 수 있습니다. 진단에 있어서 레트 증후군과 유사한 증상을 보이는 엔젤만 증후군, 프라더-윌리 증후군, 아미노산 및 유기산 대사이상, 미토콘드리아 장애, 자폐 및 뇌성마비 등과 구별을 위하여 혈액, 소변, 염색체, 뇌 영상(CT 촬영 등), 뇌파 등의 검사가 필요합니다.
레트 증후군의 모든 아동들이 이 증상 모두를 보이는 것은 아니며, 개개의 증상은 그 심각성이 다를 수 있습니다. 보통 MECP2 유전자의 돌연변이 여부 검사를 통해 확진합니다.
- 치료 및 예후
-
완치할 수 있는 방법은 없으나
현재의 발달 상황을 보존하기 위한 재활치료는 꾸준히 하는 것이 중요하며 뇌전증이 합병되었을 때는 적절한 항경련제의 복용이 필요합니다. 심부정맥 및 척추 측만, 수면 장애, 이갈기, 과호흡에 따른 복부 팽만 등의 합병증이 예상되므로 전문의와 상의하여 이에 관한 적절한 추적 관찰이 필요하다.
참고동영상)
취약X증후군 (Fragile X syndrome )
- 개요
-
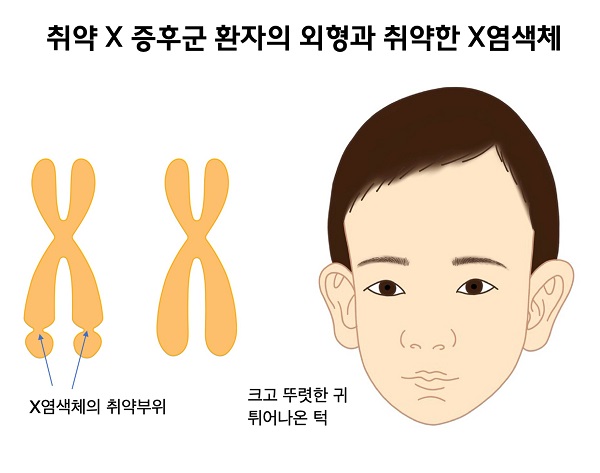
취약X증후군(Fragile X syndrome)은 학습능력과 인지기능 저하, 발달지연을 특징으로 하는 희귀질환입니다.
1943년 영국의 의사인 Martin과 Bell이 X-연관열성유전방식으로 유전되는 정신지연의 가계를 보고한 이래 Martin-Bell증후군이라고도 불립니다. 이후 1969년 Lubs가 X 염색체에서 취약부위를 처음 관찰하였고 1977년 Sutherland가 특수한 염색체 검사로 X 염색체 장완의 끝, 정확히 Xq27-q28 부위가 끊어져 보임을 밝힘으로써 여린X 증후군이 X-연관열성유전방식으로 유전됨을 확인하였습니다. 그러나 전형적 유전방식을 따르지 않으며 무증상의 남자(Normal Transmitting Male; NTM)를 통해 보인자인 딸을 거쳐 대를 거듭할수록 임상증상이 심해지고 뚜렷해지는 양상을 보입니다.
발생빈도는 민족에 따른 큰 차이가 없으며, 미국에서는 남자 4,000명 당 1명, 여자 8,000명 당 1명꼴의 빈도를 보입니다.
보인자는 남자가 1/1,000. 여자가 1/250 빈도를 보인다고 알려져 있습니다. 2013년, 김 등은 한국에서 보인자의 빈도는 1/781 이라고 보고 하였습니다. 지능저하 남성의 약 30%, 여성의 약 10%에서 이 증후군이 발견됩니다. 정신지연 및 발달장애와 더불어 자폐적 행동양식 또는 과다한 행동 등을 보여 소아정신과 의사에게 진료를 받는 경우가 흔합니다. 국외의 다른 보고들에 의하면 자폐증 남아의 5%~15%, 정신지연 남아의 6.3%~13.6%에서 세포유전학적 검사에 근거해서 취약X증후군을 진단할 수 있다고 합니다.
1990년대 초반 Verkerk, Yu, Fu 등의 취약X증후군 연구자들은 X 염색체 장완의 27.3 부위(Xq27.3)에 위치한 FMR1(fragile X mental retardation 1)유전자의 삼핵산 CGG라는 DNA 염기서열의 반복횟수가 비정상적으로 증폭됨으로써 이 질환이 초래됨을 보고하였습니다(삼핵산반복질환). 정상인은 반복횟수가 6~50회 정도에서 정규 분포를 이루는 반면, 보인자는 불완전 돌연변이(전구변이: premutation)로 대개 임상증상이 없는 여자의 경우 55~200회로 삼핵산 반복서열이 약간 증가하고, 임상증상이 있는 완전 돌연변이(완전변이: full mutation)의 경우 삼핵산 반복서열이 230회 이상입니다. 일반적으로 200~2,000회 이상 삼핵산이 증가하면 임상적으로 지능저하를 동반한 전형적인 취약X증후군이 초래됩니다.
- 증상 및 징후
-
머리는 큰 경향을 보이고, 귀모양이 크며 반복적인 중이염에 걸리기도 합니다.
근육긴장저하증과 경한 운동지연이 다소 흔합니다. 위식도역류에 의한 구역 증상을 보일 수 있습니다. 남성환자는 중등도의 여성환자는 경도의 지능저하를 보입니다. 가끔씩 간질발작과 뇌파의 이상을 보입니다. 뇌 MRI에서 뇌실주변의 이소성(periventricular heteropia)을 보이기도 합니다.
<출처 : 질병관리청 헬프라인 희귀질환정보>
환자의 행동장애로는 주의력결핍 과잉행동장애(ADHD), 적대적 반항장애, 유뇨증, 유분증이 있습니다.
그 외에도 공격성, 불안, 자폐증 양상 등이 나타날 수 있습니다. 또한 돌발적이고 잠시도 앉아있을 수 없는 행동과잉, 집중력의 문제, 일정시간 동안 한 가지 게임이나 문제를 지속해서 풀 수가 없습니다. 또한 자폐증 유사행동으로 손 흔들기, 손 물어뜯기, 눈 접촉을 잘 하지 않음, 부끄러움 등의 태도를 보입니다. 안과적 문제로 사시, 치과적 문제로 교합장애(malocclusion), 심장질환으로 승모판탈출증(mitral valve prolapse)을 보일 수 있습니다. 비뇨생식기 기형으로는 거대고환(macroorchidism)으로 고환이 9세 경에 두드러지게 커지기 시작하고 사춘기에 그 크기가 지속적으로 증가하여 14세까지는 고환의 평균 용적이 50cc에 이르게 됩니다.
근골격계 문제로는 관절의 과신전성(hyperextensibility)이 특징인데 손가락관절 과신전, 편평발(pes planus), 관절탈구, 곤봉발(club foot), 척추측만증 등을 보일 수 있습니다.
일반적으로, 여성은 부모로부터 X염색체를 각각 하나씩 받아서 X염색체가 2개입니다. 만일 X염색체 하나가 취약하더라도 나머지 정상 X염색체가 취약 부분을 상쇄할 수 있기 때문에 여성에서 취약X증후군이 발생하면 남성에 비해 증상이 심하지 않고 외양도 심하지 않습니다. 여성환자의 절반가량은 증상이 없고 나머지 절반은 학습 장애, 행동 장애 혹은 지능 장애가 나타나는 정도입니다. 남성은 어머니로부터 X 염색체를 받고 아버지로부터 Y염색체를 받습니다. 어머니의 취약한 X 염색체가 유전되면 X염색체를 상쇄해 줄 유전 정보가 Y염색체에는 없기 때문에 여성에 비해 증상이 심하고 외양도 분명하게 나타납니다.
- 진단
-
과잉행동, 지능저하, 안면기형 등의 소견과 염색체 검사나 FMR1 유전자 검사를 통해 진단하는데
환자 혈액에서 DNA를 추출하여 Xq27.3 부위 DNA의 삼핵산 반복서열이 증가(trinuleotide repeat expansion)되어 있는 분절을 증명합니다. 정상인에 비해 증가한 반복서열을 확인하는데는 비교유전자교잡법 (comparative genomic hybridization, CGH)등이 도움이 됩니다.
취약X증후군의 유전자 검사를 위한 적응증은 다음과 같습니다.
- 원인불명의 지능저하, 발달지연, 학습장애를 가진 남녀
- 자폐증과 유사한 증상을 가진 남녀
- 진단되지 않은 지능장애 또는 여린엑스 증후군의 가족력이 있는 개인
- 과거 염색체검사에서 정상이었거나 결론을 내릴 수 없게 된 경우
- 보인자인 산모의 태아 등
산전진단
유전성 정신지연의 가족력이 있는 가족구성원에서 말초혈액의 유전자검사를 시행하여 보인자 여부를 확인합니다. 보인자의 경우 수정란, 융모막, 양막세포에서 착상 전 또는 산전 유전자검사를 시행하여 수정란 또는 태아가 이환 되었는지 정확히 진단할 수 있습니다.
- 치료 및 예후
-
발생할 수 있는 개별 증상에 대한 예방과 관리가 필요합니다.
환아의 지능저하는 교육에 의해 어느 정도 극복될 수 있다는 점에서 조기중재가 매우 중요합니다. 안과적 검진으로 사시유무를 검진하며, 이비인후과적 진료로는 중이염이나 청력장애 유무를 검진합니다. 심잡음이 들린다면 심초음파 검사를 하여 승모판의 부전증이 있는지 검사합니다. 증상이 심하거나 승모판 역류가 동반되었을 경우 예방적 항생제를 수술이나 치과적 시술 전에 투약해야 합니다. 성장과 발달의 정기적 점검이 필요한데 특히 행동장애 유무를 면밀히 조사해야 합니다. 의학적 합병증이 동반될 경우 적절한 약물과 외과적 치료를 시행하며 행동수정요법 등의 특수한 교육이 요구됩니다.
주의력 결핍 과잉행동장애의 치료
행동교정(Behavioral intervention)과 개별화된 치료(Individualized therapy)에 더불어 중추신경 자극제(CNS stimulant)의 사용이 취약X증후군 환자의 주의력 결핍 과다활동장애 증상의 호전을 가져옵니다. 그러나 5세 이하의 환아에 대해서는 자극제의 사용이 불안감 및 다른 행동장애를 야기할 수 있으므로 클로니딘(Clonidine)과 구안파신(Guanfacine)과 같은 알파자극제(alpha adrenergic receptor agonist)의 사용이 바람직합니다.
불안에 대한 치료제로 선택적 세로토닌 수용체 억제제(Selective serotonin reuptake inhibitors)의 사용이 일반적이며 공격성과 기분변동에 대한 항정신약물의 사용이 도움을 줄 수 있습니다.
조기에 발견하면 전반적으로 예후는 좋습니다.
여성의 절반가량과 거의 모든 남성에서 지적장애가 나타납니다. 이 질병을 가진 환자의 수명은 대체로 정상에 가깝습니다. 전구돌연변이(premutation)를 가진 사람들에서 증상이 나타날 수 있습니다. 이들 중 여성의 1~5%는 조기난소기능저하증을 경험하며 남성과 여성 모두에서 여린 엑스 진전-실조 증후군(fragil X tremor-ataxia syndrome, FXTAS)이라는 신경 질환이 발생할 가능성이 높습니다.
출처 : 질병관리청 헬프라인 희귀질환정보
신경섬유종증 (Neurofibromatosis )
- 개요
-
신경섬유종증 (Neurofibromatosis, NF)은 신경계에서 종양이 자라게 되는 유전 질환입니다.
종양은 신경을 구성하는 세포와 신경을 감싸고 보호하는 얇은 막인 수초에서 시작됩니다. 종양은 말초신경에서 주로 발생하지만 드물게 뇌와 척수에도 생기며 주변 조직을 변형시키거나 기능을 떨어뜨리기도 합니다.
상염색체 우성 양식으로 유전하는 유전 질환으로, 신경섬유종 제1형은 17번 염색체(17q 11.2)에 존재하는 NF1 유전자 결함, 그리고 제2형의 경우 22번 염색체 장완(22q11.2)에 존재하는 NF2 유전자의 결함에 의해 발생합니다. 신경섬유종증 환자의 약 50%는 가족력 없이 새로운 돌연변이에 의해 발생됩니다. 한쪽 부모가 질환이 있을 경우 50%의 확률로 자녀에게 유전되며 전체 환자 중 약 50%는 이렇게 부모에게 유전된 경우입니다. 나머지 절반의 환자는 자연발생(유전에 의하지 않은) 유전자 돌연변이에 의한 것이므로 가족 병력이 없습니다.
- 증상 및 징후
-
신경섬유종증에는 세 가지 형태가 있습니다.
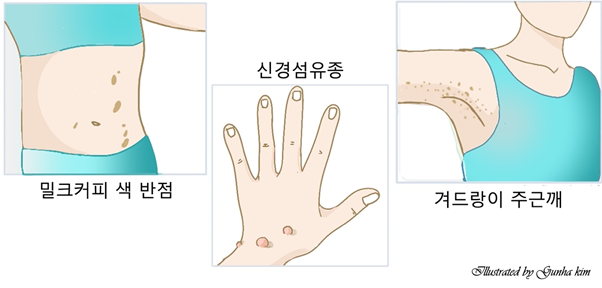
1형 신경섬유종증(NF1)은 그 중 가장 흔한 형태의 질환이며 증상은 환자마다 다를 수 있습니다. 소아에서 일반적으로 볼 수 있는 가장 초기의 임상 소견은 밀크 커피색 반점(카페오레 반점, cafe-au-lait-spot)입니다. 이들은 태어날 때부터 있을 수도 있고 시간이 지나면서 나타날 수도 있으며, 어린 시절 내내 크기, 숫자 및 색소침착이 증가합니다. 사춘기 이전에는 최대 직경이 5mm 이상, 사춘기 이후에는 최대 직경이 15mm 이상인 반점이 6개이상 관찰되면 의미가 있습니다. 주근깨는 출생 시에는 거의 나타나지 않고 3-4세 이후에 직경 2-3mm의 과도색소침착이 겨드랑이 또는 서혜부에서 다수 관찰되며, 목이나 몸 및 사지에도 나타날 수 있습니다. 피부 혹은 피하 신경섬유종은 고무처럼 탄성이 있고 자주빛을 띤 병변으로 크기는 수 mm에서 수 cm까지 다양하며 모양은 편평하기도 하고 결절처럼 나타나기도 합니다. 어린 소아에서는 드물게 나타나지만 시간이 지남에 따라 나이가 많은 소아, 청소년 및 성인에서 나타나기 시작합니다. 얼기형 신경섬유종은 신경줄기가 광범위하게 굵어진 것으로 출생 시부터 관찰되며, 얼굴의 안와 및 측두 부위에 호발합니다. 2개이상의 신경섬유종이나 하나 이상의 얼기형 신경섬유종이 관찰되면 의미가 있습니다. 안과적 양상으로는 Lisch 결절, 녹내장, 시각신경 교종(optic glioma) 등이 있습니다. Lisch 결절은 홍채의 과오종으로 6세경 나타나기 시작하여 성인이 되면 95%에서 관찰됩니다. 세극등(slit lamp) 검사로 관찰할 수 있으며, 2개 이상 관찰되면 의미가 있다. 시각신경 교종은 신경섬유종증에서 가장 흔히 발생하는 중추신경계 종양으로, 환자의 15%에서 관찰되며, 대부분 무증상이나 크기가 커지면 시력 저하나 시야 감소 등 시작 증상이 나타나며 시상하부를 침범하면 내분비 증상을 초래할 수도 있습니다. 점차 커질 수 있으므로 10세 이전에는 매년 안과검사가 필요합니다.
대표적 신체 징후는 다음과 같습니다.
* 피부의 밀크 커피색 반점(카페오레 반점, cafe-au-lait-spot)
* 겨드랑이 혹은 사타구니 부위의 주근깨
* 분리형 신경섬유종(discrete neurofibroma) 또한 얼기형 신경섬유종(plexiform neurofibroma)
* 리쉬 결절(Lisch nodule)이라고 불리는 눈의 홍채에 있는 반점
* 특징적인 뼈의 변화 (정강뼈가 휘거나 안면 비대칭, 척추측만증)
* 시신경 교종
[글/그림: 김건하 한국원자력의학원 원자력병원(Korea cancer center hospital) 소아청소년과 신경발달클리닉]
1형 신경섬유종증은 대부분 양성종양이지만, 드문 경우에는 악성으로 진행될 수 있으며 신경이 있는 신체 모든 부위에 발생할 수 있습니다. 이 밖에도 저신장, 대두증, 학습장애, 경련, 성조숙증, 이차성 고혈압(신혈관협착이나 부신종양이 원인) 등이 동반될 수 있습니다.
다음으로, 2형 신경섬유종증(NF2)은 제8 뇌신경의 전정신경 가지에서 전정신경초종(vestibular schwannoma)이라는 종양이 발생하는 질환입니다. 과거에 청신경종양(acoustic neuroma)으로 불렸으나, 실제로는 전정신경에서 발생하여 인접한 청신경을 누르면서 증상을 일으키는 것이 확인되어 전정신경초종으로 불리게 되었습니다. 소아기에서는 미세한 피부 신경초종이나 눈의 렌즈혼탁 등이 나타날 수 있으나 간과하기 쉽습니다. 주로 청소년기 이후 전정신경초종이 발생하면서 난청, 이명, 그리고 균형 장애가 경미하게 나타나면서 진단되고 계속 크기가 커져서 청신경을 누르기 시작하면 서서히 청력도 떨어지기 시작합니다.
세번째 형태인 신경초종증(schwannomatosis)은 신체의 여러 곳에서 다발성 신경초종이 발생하는 아주 드문 질환입니다. 청신경을 제외한 신체 거의 모든 신경에 양성 종양이 발생할 수 있습니다. 주 증상은 신경초종이 신경이나 인접 조직을 압박하면서 발생하는 통증이며 손가락과 발가락에 무감각, 따끔거림 또는 쇠약이 생길 수도 있습니다.
- 진단
-
대부분 일상적인 검진을 받다가 밀크커피색 반점을 발견하거나, 미용과 관련된 증상 때문에 의사 진료를 받거나, 또는 신경섬유종증에 대한 가족력이 있는 경우 신경섬유종증에 대한 진단검사를 받게 됩니다. 의사는 진단기준에 포함된 임상기준들을 하나씩 확인한 후 최종 진단을 내리게 되며 이를 위해 가족력확인, 진찰, 영상검사, 안과 협진 및 유전자 검사 등을 시행하게 됩니다.
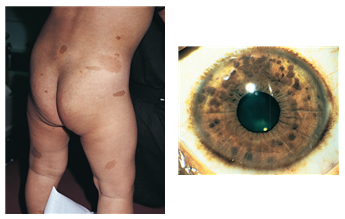
<밀크커피색반점과 Lisch 소결절; 출처 Nelson Textbook of Pediatrics, 21th ed, p3141, 3476>
- 치료 및 예후
-
제1형 신경섬유종증은 정기적인 검사를 통해 합병증을 조기에 발견하는 것이 중요합니다.
매 방문마다 피부를 확인하고 뼈의 이상 유무와 고혈압 발생 유무를 확인 합니다. 척수 신경섬유종의 증상은 미묘하고 천천히 진행될 수 있으므로, 근육 위축이나 감각이상, 쇠약 등 감각/운동 증상의 미묘한 변화를 주의 깊게 확인해야 합니다. 특히, 시신경 병변의 조기 발견을 위해서는 매년 안과 검진이 중요하며 시신경교종 발생 부위에 따라 성조숙증이 나타날 수 있으므로 성성숙도를 확인해야 합니다. 또한, 발달지연 및 학습장애가 동반되는 경우가 종종 있으므로 조기 개입을 위해 발달 속도 및 학업성취도에 대한 감시가 필요합니다. 한편, 분리형 신경섬유종은 미용적 또는 의학적 이유로 제거하기도 하지만, 신경섬유종의 한 형태인 얼기형 신경섬유종은 건강한 정상세포 주변에 얽혀있기 때문에 기능적 손상 없이 완벽하게 제거하는 것은 매우 어렵습니다. 다행히도, 이런 경우에 차선책으로 쓸 수 있는 첫 신경섬유종증 치료제가 2020년 미국 식품의약국에서 승인되었으며 2021년 국내에서도 긴급 승인되어 사용이 가능하게 되었습니다.
2형 신경섬유종증의 종양은 환자연령, 종양성장 속도에 따라 수술여부를 선택합니다.
일반적으로 수술 후 청력이 감소될 수 있는 위험이 있어 종양으로 인한 청력 저하가 어느 정도 진행될 때까지 수술을 미루게 됩니다. 또한 감마나이프 등을 활용한 방사선치료는 수술보다 신경손상의 위험은 낮지만 큰 종양을 축소하는 데는 효과가 충분하지 않을 수 있습니다. 혈관을 표적으로 하는 약물을 사용한 화학요법은 종양의 크기를 줄이고 청력을 개선할 수 있지만 일부는 전혀 반응하지 않거나 일시적으로만 반응할 수 있습니다.
1형 신경섬유종증의 증상은 대부분은 경미하여 정상적인 사회활동이 가능합니다.
그러나 신경섬유종으로 인한 미용 및 심리적 문제가 동반되기도 합니다. 일부에서 학습장애, 뇌전증, 양성종양으로 인한 신경계 합병증 및 악성종양이 발생하여 치료가 필요합니다. 2형 신경섬유종증의 경과는 개인마다 크게 다르지만 양쪽 귀의 청력 상실은 대부분의 환자에서 발생합니다.
결절성경화증 (Tuberous sclerosis)
- 개요
-
결절성 경화증(tuberous sclerosis)은 정신지연, 뇌전증, 피부병변 등의 증상이 특징적으로 나타나고 6,000-9,000명 당 한 명의 빈도로 드물게 발생하는 선천성 질환입니다.
관련 증상 역시 워낙 복잡하다보니 소아신경과에서부터 소아정신과, 소아심장과, 소아신장과, 안과, 피부과, 성형외과, 소아치과 그리고 유전학과에 이르기까지 다양한 분야의 전문가들이 총 동원되어야하는 어려운 질환입니다. 이 병과 관계있는 TSC1, TSC2 유전자는 상염색체 우성으로 유전되는데, 환자의 1/3정도는 부모에게서 질병을 물려받고, 나머지는 새로운 유전자 돌연변이에 의해 발생합니다. 두 유전자 이상에서 나타나는 임상적인 발현은 비슷하나 TSC2 환자에서 신경학적인 이상이 더 많다고 알려져 있습니다
중추신경계 및 다양한 신체 부위를 침범하여 여러 가지 증상을 일으키며, 종양을 일으킬 수 있기 때문에 장기적인 관찰이 필요한 병입니다.
- 증상 및 징후
-
증상의 발현 연령, 중증도, 진행 속도, 임상 양상이 환자에 따라 다양하게 나타나며, 4개의 주 증상은 정신지연, 뇌전증, 피부병변, 신체 각 부위의 종양입니다.
정신지연의 빈도는 다양한데, 일부의 환자에서는 원인을 알 수 없지만 자폐적인 성향을 보입니다. 1/3의 환자에서는 다른 임상적 소견으로 결절성 경화증으로 진단되지만 정상적인 지능을 갖습니다. 최근의 연구에서 55%의 환자들은 지능지수가 70 이상이며 30.5%의 환자들은 21 이하의 지능지수인 것으로 보고되었습니다. 처음에는 정상적인 지능을 보이다가 8-14세 이후에 지능저하가 나타나는 경우도 있는데, 이런 경우는 간질이 조절되지 않는 것에 영향을 받았거나 뇌종양에 의해 뇌압이 증가되어 나타나는 것으로 보입니다. 전반적인 지능의 문제 이외에 환자들은 행동상의 문제나 자폐적 성향과 같은 정신과적인 문제, 학습장애 등을 보일 수도 있습니다.
뇌전증증은 가장 흔한 증상입니다. 나이가 어린 영아에서는 주로 영아연축의 뇌전증형태를 보이고 나이가 듦에 따라 점차 부분발작이나 전신발작을 보일 수 있습니다. 뇌전증은 아주 빠른 경우 생후 1주일부터 발생할 수도 있는데, 뇌전증이 일찍 발생할수록 정신지연의 가능성이 많습니다.
피지샘종의 가장 특징적인 피부소견으로 붉고 구진성의 발진이 코, 턱, 뺨, 광대뼈 등의 부위에 1에서 5세 사이에 나타나기 시작하고 점차 크기와 숫자가 증가합니다. 색소탈실반은 다양한 크기로 백반증과 닮았는데, 체간이나 사지에 타원형이면서 불분명한 경계부를 보입니다. 대개 피지샘종보다 일찍 나타납니다. 다른 피부 병변으로는 손톱주위의 섬유종, 샤그린반점, 담갈색반점 등이 있습니다. 섬유종은 체간이나 잇몸 주위에도 나타날 수 있습니다. 일부의 환자들은 부분적인 흰머리 다발소견이 보이는데, 이것은 이 질환의 아주 초기 소견으로 나타나기도 합니다.
뇌의 병리학적 소견으로는 뇌의 피질과 뇌실막 밑에 신경세포의 숫자는 감소하고 과다증식된 성상세포와 크고 다핵의 기형적인 세포로 구성된 결절이 관찰되는 것이 특징입니다.
50% 정도의 환자에서 망막 종양이나 망막의 다른 이상이 동반됩니다. 종양이 흔한 다른 부위로는 피부, 폐, 뼈, 간, 비장이 있습니다. 신경계 이외의 증상은 대개 성인기에 발생하게 됩니다. 콩팥의 질환이 환자의 건강에 영향을 미칠 수 있습니다. 3%-5%의 환자에서 보이는 다낭콩팥병을 가진 환자들은 신부전이 진행하여 신장이식이 필요하게 되기도 합니다. 좀 더 흔하게 보이는, 콩팥에 하나 혹은 여러 개의 콩팥낭이 있는 경우는 대개 신부전은 발생하지 않고 혈관근육지방종과 관련이 있을 수 있습니다. 폐에 발생하는 혈관근육지방종은 치명적일 수 있으며 거의 여자 환자들에게서 발생합니다. 좌심실 내 횡문근종, 울혈성 심부전이나 부정맥도 발생할 수 있습니다.
- 진단
-
진단은 대개 특징적인 피부소견, 간질, 지적장애 등에 근거하여 이루어집니다. 영아의 경우에는 피부의 탈색소반, 영아연축, 발달지연이 진단의 주요소견입니다. 얼굴의 피지샘종, 손톱주위의 섬유종, 뇌겉질의 결절이나 뇌실막 밑의 과오종, 안저의 다발 과오종 중 하나 이상의 소견을 가지거나 영아연축, 색소탈실반, 샤그린반점, 안저의 과오종, 뇌실 내 또는 뇌실 주변의 결절석회침착, 양쪽 신장의 혈관근육지방종, 심장의 횡문근종 중 2가지 이상의 소견을 보이면 결절성 경화증의 진단이 가능합니다.
뇌 CT 및 MRI
뇌 CT 스캔 검사로 뇌의 결절석회침착을 쉽게 관찰할 수 있으나 3-4세가 지나야 이러한 소견이 나타나는 수도 있습니다. 뇌 MRI는 결절성 경화증을 확진하는데 가장 확실한 방법으로 90% 이상의 환자들이 뇌 MRI 소견 이상을 보이는데, 뇌겉질의 결절, 과오종 및 종양의 소견 이외에도 뇌백질의 이주 장애, 뇌량의 형성이상 등의 소견을 보여 진단에 유용하게 이용됩니다.
유전자 검사
결절성 경화증의 경우 진단은 임상적 기준에 의해 진단하므로 유전자 검사로 확진하는 것은 아닙니다. TSC1, TSC2 유전자에 대한 검사는 2002년 이후로 가능하게 되어 있으므로 임상양상만으로 진단이 불명확한 경우 검사해 볼 수 있으나, 2/3의 환자에서 산발적으로 발생하거나 섞임증 등에 의해서도 발생할 수 있으므로 모든 환자에게 일률적으로 검사할 필요는 없습니다.
기타
복부 초음파, 심장 초음파 및 심전도, 안과 진찰, 피부과 진찰, 신경발달검사를 통하여 연관된 기관의 이상을 찾아낼 수 있습니다.
- 치료 및 예후
-
이 병을 완치시키는 방법은 현재까지는 없고 주된 치료는 여러 가지 나타나는 증상들에 대한 대증적 치료입니다.
뇌전증이 발생한 경우 항경련제를 사용합니다. 영아연축이 있는 결절성 경화증 환자의 간질은 Vigabarin이 가장 효과적인 것으로 알려져 있습니다. 행동장애와 지능장애가 있는 경우에는 개개인의 환자에게 적절한 약물을 사용하거나 특수교육, 작업치료 등을 받는 것이 도움이 됩니다. 피부병변에 대한 레이저 치료나 기타 수술적 치료를 고려해 볼 수 있습니다.
종양의 발생 시 외과적 치료를 고려합니다. 뇌 내 병변의 경우 뇌겉질의 결절이 하나 있는 경우에는 환자에 따라 수술로 제거해 주는 것이 도움이 될 수도 있습니다. 뇌실 내 종양이 있는 소아 환자의 경우 뇌척수액 통로를 막는 합병증이 발생할 때까지 그냥 관찰하는 것이 좋은지 미리 치료하는 것이 좋은지에 대한 협의는 아직 이루어지지 않았습니다.
결절성 경화증은 평생 동안 지속되는 상황이며 병의 합병증으로 심장, 뇌, 신장 등에 종양이 발생할 수 있으므로 환자는 주기적으로 전문의의 검진을 받으며 종양이 발생하는지 계속 관찰하는 것이 필요합니다. 이 병으로 사망할 수 있는 주요 원인으로는 간질중첩증, 콩팥의 질환, 뇌종양, 폐의 림프종 등입니다.
출처 : 질병관리청 헬프라인 희귀질환정보
척수성근위축증 (Spinal muscular atrophy)
- 개요
-
척수성 근위축증 (Spinal muscular atrophy, SMA) 은 운동신경세포의 기능손상으로 인해 근력이 저하되고, 근육이 위축이 되는 퇴행성 신경질환입니다. 영유아부터 성인기에 이르기까지 다양한 시기에 증상이 나타날 수 있고 아형에 따라 기기, 걷기, 머리와 목 가누기, 삼키기 등 운동 기능에 영향을 줄 수 있습니다. 발병률은 대략 신생아 6,000~10,000 명당 1명 이고, 여성과 남성에서 동일한 비율로 나타납니다.
이 질환은 5 번 염색체에 있는 SMN1 유전자의 돌연변이에 의해 발생합니다.
SMN1 유전자는 SMN 단백질을 합성합니다. 척수성 근위축증에서는 SMN1 유전자의 돌연변이에 의해 SMN 단백질의 결핍이 발생하고 이로 인해 근육과 운동 신경세포 사이의 신호전달에 문제가 생기면서 근육이 적절한 기능을 하지 못하고 방치되며, 근력저하, 근위축이 생기게 됩니다. 이 질환은 상염색체 열성 유전 질환으로 두 개의 SMN1 유전자의 돌연변이가 질환을 일으킵니다. 돌연변이가 한 개만 있는 경우는 “보인자” 라고 하며, 이런 경우에는 증상이 없습니다. 대부분의 경우는 보인자인 부모로부터 한 개씩 돌연변이 유전자를 받아서 발생합니다. 보인자 빈도는 약 40 에서 60 명중 1명으로 보고되고 있습니다. 만약 부모가 모두 보인자인 경우에는 척수성 근위축증 아이가 출생할 확률은 25% 가 됩니다.
5 번 염색체에는 SMN1 유전자와 유사한 SMN2 유전자가 있고, 사람마다 가지고 있는 SMN2 유전자의 개수는 다양합니다. SMN2 유전자는 SMN 단백질을 약 10% 정도 합성할 수 있습니다. 이 때문에 SMN2 유전자의 개수는 건강한 사람은 관계 없지만, 척수성 근위축증에서 SMN2 유전자가 많은 사람은 일반적으로 적은 사람보다 덜 심각한 형태의 질환을 보이게 됩니다.
- 증상 및 징후
-
척수성 근위축증의 아형, 질환의 단계, 개인차에 따라 그 증상은 다릅니다.
일반적으로 대칭적으로 근력이 떨어지고, 사지의 근위부에서 더 광범위 합니다. 근긴장도 저하, 얼굴 근육 기능저하, 혀 근육 수축, 호흡기능 저하가 나타나게 됩니다. 심부건 반사는 거의 없거나 미약합니다. 지능 저하나 괄약근 침범은 없습니다. 질환이 진행되면 관절 구축이나 척추측만증이 동반되기도 합니다.
척수성 근위축증은 발병 시기, 획득 가능 운동 발달 정도에 따라 제 1 형에서 제 3 형으로 분류 됩니다. 최근에는 성인에서 발병하는 4형이 추가되었습니다. 약 4분의 1이 1형이고, 2분의 1정도가 2형이며 나머지가 3형에 속합니다.
제 1형
가장 흔하게 발생하고 증상이 가장 심한 형태로 척수성 근위축증으로 진단받은 환자의 약 50%를 차지합니다. 출생 후 6 개월 미만에서 증상이 나타납니다. 도움 없이 앉는 것이 불가능하며, 근긴장저하, 머리 제어 불가능, 안면근력 약화, 호흡곤란등의 증상을 보입니다. 연수 침범으로 혀의 근다발수축을 보이며 빠는 힘이 약하고 잘 못 삼킵니다. 이로 인해 기도 보호가 안되어 흡인성 폐렴이 발생할 위험이 큽니다. 치료를 하지 않으면 대부분 2 세 이전에 호흡 부전으로 사망합니다.

[그림: “소아신경학, 제3판” 대한소아신경학회, 군자출판사, 2021 ]
제2형
중간 정도의 심한 증상을 특징으로 합니다. 발병 시기는 대부분 6개월 이후 18개월 이전에 나타납니다. 혼자 앉는 것은 가능하나 독립 보행이 불가능하고, 휠체어가 필요합니다. 진행되면 관절 구축이나 척추측만증을 보일 수 있습니다. 삼키는 힘이 약하여 체중증가가 잘 안될 수 있으므로 비위관 영양이 필요합니다. 상기도 감염 시 근약화로 기도내 분비물 배출과 기침을 하기 어려울 수 있습니다. 청소년기와 젊은 성인기에 호흡부전으로 흔히 사망합니다.
제3형
3형 척수성 근위축증은 증상이 다양합니다. 근력 약화는 생후 18 개월 이상에서 나타납니다. 독립 보행이 가능하나, 점차적으로 근력이 약화되어 보행 능력을 잃게 되고 휠체어 등의 도움이 필요합니다. 척추측만증이 발생하나 2형보다 늦은 나이에 발생합니다.
제4형
가장 드문 아형입니다. 성인기에 증상이 나타나며 가장 경한 운동 증상을 보입니다. 보행이 가능하며, 호흡, 식이 문제가 없습니다. 1형이나 2형과 달리 관절운동제한이 상대적으로 흔합니다.
- 진단
-
진단은 유전자 검사를 통해 할 수 있으며, 그 외 근전도 검사 및 근육 조직검사를 시행하기도 합니다.
척수성 근위축증은 근전도 검사에서 신경성 패턴을 나타내고, 근육 조직 검사에서 근육에 퇴행성 위축이 나타나 있는 것을 확인 할 수 있습니다. 최근에는 척수성 근위축증이 의심된다면 근전도 검사, 근육 조직 검사 없이 첫 단계의 유전자 검사로 95% 의 환자를 확진할 수 있기 때문에 유전자 검사 하나만 시행하여 진단하기도 합니다.
- 치료 및 예후
-
이전에는 각종 부작용과 합병증을 조절하고 완화시키기 위한 대증요법과 지지요법치료를 중점적으로 하였으나, 최근에는 다양한 치료제들이 개발되어 환자들의 생존률과 운동기능을 향상시키는 것이 가능해졌습니다.
SMN1 유전자를 직접 주입하는 치료와, SMN2 유전자에서 SMN 단백질의 합성을 증가시키는 치료가 있습니다. 치료 후에는 치료 시기, 질환의 아형에 따라서 운동 능력, 호흡기능, 식이섭취기능 등이 향상 또는 유지 될 수 있게 됩니다. 조기에 치료할 수록 생존하는 운동신경세포 수가 많아지고 그 기능을 유지할 수 있어서 더 좋은 예후를 기대할 수 있습니다. 그러나 이 치료제들로 척수성 근위축증이 완치가 되는 것은 아닙니다. 질환의 어느 시점에 치료를 시작 했는가에 따라 근력약화 증상이 남아있을 수 있습니다. 또한 호흡 기능, 식이섭취기능, 관절 가동성을 유지하고 근골격계의 변형을 예방하기 위해서는 지속적인 관리가 필요합니다. 근육 위축에 의한 형태 변형과 기능 장애를 줄이기 위해 물리치료, 보행이나 기립을 위한 보조 기구를 이용할 수 있습니다. 호흡과 관련된 근육이 매우 약해졌을 경우 인공호흡기를 사용하여 호흡을 보조 할 수 있습니다. 연하곤란 발생 여부를 모니터링을 하고, 필요하다면 장관 영양을 통해 영양 공급을 해야합니다.